Post by Olga Golosova on Nov 9th, 2018 at 3:07pm
Hi Julio.
Quote:
Thank you for your feedback! Apparently, we should simplify this feature in UGENE and improve documentation. :)
First of all, note that you shouldn't use "Map Sanger reads to reference" tool. It implies that you provide the Sanger reads in *.ab1 or similar format (see a video about this feature).
For your task there are a couple of workarounds you may try first. Please try them and write, if it works for you or not.
1) If you have a few sequences you would like to align to the reference sequence:
Note that you should manually convert a read sequence to its reverse-complement sequence in this case, if required. In the Sequence View select "Edit > Reverse-complement sequence".
2) Sometimes when you map a lot of reads/contigs to a reference sequence, BWA-MEM helps, even if originally it is used for NGS reads:
BWA-MEM automatically revert a read sequence, if required.
The result is opened in the Assembly Browser.
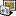 option1_open_reference_as_alignment.png (54 KB | 344
)
option1_open_reference_as_alignment.png (54 KB | 344
)
option1_align_reads_to_reference.png (55 KB | 340
)
option2_bwa-mem_settings.png (105 KB | 388
)
Quote:
|
Thank you for your feedback! Apparently, we should simplify this feature in UGENE and improve documentation. :)
First of all, note that you shouldn't use "Map Sanger reads to reference" tool. It implies that you provide the Sanger reads in *.ab1 or similar format (see a video about this feature).
For your task there are a couple of workarounds you may try first. Please try them and write, if it works for you or not.
1) If you have a few sequences you would like to align to the reference sequence:
- Export the reference sequence to an alignment format to open it in the Alignment Editor. Select "Export/Import > Export sequences as alignment" in the context menu for the sequence object in the Project View tp do this (see "option1_open_reference_as_alignment.png").
- Then make sure the Alignment Editor window is opened and click "Align sequence to this alignment" button on the toolbar (see "option1_align_reads_to_reference.png" and documentation).
Note that you should manually convert a read sequence to its reverse-complement sequence in this case, if required. In the Sequence View select "Edit > Reverse-complement sequence".
2) Sometimes when you map a lot of reads/contigs to a reference sequence, BWA-MEM helps, even if originally it is used for NGS reads:
- Select "Tools > NGS data analysis > Map reads to reference" in the main UGENE menu.
- Set "Mapping tool" to BWA-MEM, input the reference sequence and the reads, click "Start" ("option2_bwa-mem_settings.png").
BWA-MEM automatically revert a read sequence, if required.
The result is opened in the Assembly Browser.
option1_open_reference_as_alignment.png (54 KB | 344
) option1_align_reads_to_reference.png (55 KB | 340
)
option1_align_reads_to_reference.png (55 KB | 340
) option2_bwa-mem_settings.png (105 KB | 388
)
option2_bwa-mem_settings.png (105 KB | 388
)
 https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=Characiformes_mito.fasta (31 KB | 288
)
https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=Characiformes_mito.fasta (31 KB | 288
)