Post by roblogan6 on Jun 10th, 2016 at 4:34am
I have used mafft to align multifasta files with a txt extension. I made the attached work flow, trying to get one file per multifasta file, that contains the consensus sequence in fasta format for downstream analysis. I am getting an error that my input files are not in the correct format. They look something like this:
>TAGCTAGCTAGC
--TAGCTAGC--TAGC-T-TAGC----GGG
>TAGCTAGCTAGC
AGTAGCTAGCCTAGC---TAGCCT-GG-
How can I use these .txt files in fasta format, previously aligned using mafft, and UGENE to produce one file per multifasta file with a consensus sequence, such as the following (depending on the parameters used to generate consensus sequence)
>TAGCTAGCTAGC
--TAGCTAGC--TAGC---TAGC----GG
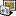 Screen_Shot_2016-06-09_at_5_33_55_PM.png (232 KB | 351
)
Screen_Shot_2016-06-09_at_5_33_55_PM.png (232 KB | 351
)
>TAGCTAGCTAGC
--TAGCTAGC--TAGC-T-TAGC----GGG
>TAGCTAGCTAGC
AGTAGCTAGCCTAGC---TAGCCT-GG-
How can I use these .txt files in fasta format, previously aligned using mafft, and UGENE to produce one file per multifasta file with a consensus sequence, such as the following (depending on the parameters used to generate consensus sequence)
>TAGCTAGCTAGC
--TAGCTAGC--TAGC---TAGC----GG
Screen_Shot_2016-06-09_at_5_33_55_PM.png (232 KB | 351
)