Post by Olga Golosova on Mar 3rd, 2015 at 9:09pm
Hi Leiga!
A:
UGENE integrates FastQC tool for getting the statistics like shown on the image. You can watch this video to learn how to interpret the FastQC reports: http://www.youtube.com/watch?v=bz93ReOv87Y
To run it in UGENE 1.16, select "Tools > NGS data analysis > Reads quality control" in the main menu.
B:
Could you please give more details? What is the goal? Wouldn't it help to map the reads to the plasmid sequences?
By the way, have you succeeded using SPAdes in UGENE (http://ugene.unipro.ru/forum/YaBB.pl?num=1422521083) or you used another de novo assembler?
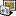 fastqc.png (102 KB | 478
)
fastqc.png (102 KB | 478
)
A:
UGENE integrates FastQC tool for getting the statistics like shown on the image. You can watch this video to learn how to interpret the FastQC reports: http://www.youtube.com/watch?v=bz93ReOv87Y
To run it in UGENE 1.16, select "Tools > NGS data analysis > Reads quality control" in the main menu.
B:
Could you please give more details? What is the goal? Wouldn't it help to map the reads to the plasmid sequences?
By the way, have you succeeded using SPAdes in UGENE (http://ugene.unipro.ru/forum/YaBB.pl?num=1422521083) or you used another de novo assembler?
fastqc.png (102 KB | 478
)