Post by Ivan Efremov on May 3rd, 2011 at 10:47am
Hi Hieu,
here are the answers:
1. You can convert fastq to fasta using "Save a copy" button in project view, or using a simple workflow "Read Sequence -> Write Fasta". After converting fastq to fasta you will be able to use the FormatDB tool.
2. You have to select that annotations from remote blast should be used in genbank writer. See attached screenshot.
Feel free to ask more questions if further help is needed.
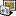 save-a-copy.png (12 KB | )
save-a-copy.png (12 KB | )
workflow-remote-blast.png (33 KB | )
here are the answers:
1. You can convert fastq to fasta using "Save a copy" button in project view, or using a simple workflow "Read Sequence -> Write Fasta". After converting fastq to fasta you will be able to use the FormatDB tool.
2. You have to select that annotations from remote blast should be used in genbank writer. See attached screenshot.
Feel free to ask more questions if further help is needed.
save-a-copy.png (12 KB | ) workflow-remote-blast.png (33 KB | )
workflow-remote-blast.png (33 KB | )
 https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=remoteBLAST.uwl (3 KB | )
https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=remoteBLAST.uwl (3 KB | )