Post by Agu on Mar 7th, 2011 at 5:53am
Hi.
Probably soon, but I think that currently you can't (at least in a direct way).
To automate tasks you should go to "Tools/Workflow designer".
Here you have an example of how to use it:
http://www.youtube.com/watch?v=s5zp8DZxNVI&feature=related
In the latest version of UGENE (1.9.1) they added an item in the workflow designer that filters annotations by name. The problem is that with this option you can extract at once all the features of a kind, like gene or CDS, but you can not extract individual genes as UGENE is not reading qualifiers (like protein_id, label, note, etc.). Nevertheless, I was thinking some ways of solving the problem. If you have a stable size (I guess you do) for the ORFs you are interested in, you could annotate the ORFs within a certain range and then use that for the alignment. I'm attaching an example schema that could work for you (and an external query that you have to use to search for the ORFs in the range you need). Let me know if you could solve it.
The schema is:
PS: make sure you complete the unset input and output files (four including the location of "ORF search.uql") and correct the ORF size in the ORF search query.
 https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=Schema2.uwl (11 KB | ) https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=ORF_search.uql (0 KB | )
https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=Schema2.uwl (11 KB | ) https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=ORF_search.uql (0 KB | )
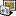 Schema.png (118 KB | )
Schema.png (118 KB | )
Probably soon, but I think that currently you can't (at least in a direct way).
To automate tasks you should go to "Tools/Workflow designer".
Here you have an example of how to use it:
http://www.youtube.com/watch?v=s5zp8DZxNVI&feature=related
In the latest version of UGENE (1.9.1) they added an item in the workflow designer that filters annotations by name. The problem is that with this option you can extract at once all the features of a kind, like gene or CDS, but you can not extract individual genes as UGENE is not reading qualifiers (like protein_id, label, note, etc.). Nevertheless, I was thinking some ways of solving the problem. If you have a stable size (I guess you do) for the ORFs you are interested in, you could annotate the ORFs within a certain range and then use that for the alignment. I'm attaching an example schema that could work for you (and an external query that you have to use to search for the ORFs in the range you need). Let me know if you could solve it.
The schema is:
- Reading several GenBank files.
- Using the external query (ORF search.uql) to search ORFs of a given size (1400 - 1600 bp in this case) and annotating them as Mitochondrion
- Filtering the annotations so only Mitochondrion is left
- Retreiven the sequences from each .gb for the Mitochondrion feature
- Saving that as multiple fasta file (just in case)
- Joining all extracted sequences into one alignment, aligning with muscle and saving the result...
PS: make sure you complete the unset input and output files (four including the location of "ORF search.uql") and correct the ORF size in the ORF search query.
https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=Schema2.uwl (11 KB | ) https://forum.ugene.net/forum/YaBB.pl?action=downloadfile;file=ORF_search.uql (0 KB | ) Schema.png (118 KB | )